Tyto vakcíny jsou podle něj „skvělým lékem“ a tvrdí, že na individuální úrovni opravdu fungují, ale jsou „nesprávnou zbraní“ pro použití v globálním měřítku, kdy jsou přítomny vysoké tlaky infekčních patogenů.
Jak tweetol 3. března:
„Jsem NESMÍRNĚ znepokojen dopadem, který budou mít současné vakcíny proti onemocnění Covid-19 při jejich stále větším použití v hromadných očkovacích kampaních, prováděných pod tlakem pandemie. Přečtěte si mé globální VAROVÁNÍ a vědecké důkazy.“
K tomuto svému varování přidává následující link: bit.ly/3q89hWZ
Obsahem dokumentu jsou slajdy z prezentace, vytvořené Dr. Bosschem, nazvané „Proč by se současné vakcíny proti onemocnění Covid-19 neměly používat k hromadné vakcinaci během pandemie?“
Znalec prostředí vakcínového průmyslu, který pracoval pro Nadaci Billa a Melindy Gatesových
Než přejdeme k prezentaci a jejímu významu, podívejme se nejprve narychlo pracovním zkušenostem Dr. Bosscheho. Dobrý popis lze nalézt na stránce Dryburgh.com, která se tomuto příběhu podrobně věnovala:
„Geert Vanden Bossche, PhD., DVM, je odborník na výzkum vakcín. Při objevech vakcín a také v předklinickém výzkumu pracoval s řadou společností a organizací, jako jsou GSK, Novartis, Solvay Biologicals a Nadace Billa a Melindy Gatesových.
Dr. Vanden Bossche také v GAVI (Globální alianci pro vakcíny a imunizaci) koordinoval očkovací program proti ebole.
Má atestaci z virologie a mikrobiologie, napsal přes 30 publikací a vynalezl patent pro aplikaci univerzálních vakcín. V současnosti pracuje jako nezávislý konzultant ve vývoji vakcín.“
Řečeno jinak, sotva dokážete najít někoho kvalifikovanějšího a zkušenějšího od toho, kdo důvěrně zná prostředí vakcínového průmyslu.
Především, Dr. Bossche je očividně provakcínový a není takzvaný „antivaxer“. Jenže i při svém provakcinačním postoji vidí nesmírná rizika a problémy hromadných očkovacích kampaní, i za předpokladu, že vakcíny fungují tak, jak mají.
Dr. Bossche varuje, že svět vytváří „nezvládnutelné monstrum“ a mění vakcíny na „biologickou zbraň hromadného ničení“
Toto je aktuální citát Dr. Bosschea:
„Člověk by dokázal vymyslet jen velmi málo jiných strategií k dosažení stejné míry účinnosti ve změně realtivně neškodného viru na biologickou zbraň hromadného ničení.“
Jak vysvětluje stránka Dryburgh.com:
Dr. Bossche je přesvědčen, že vakcinologové, kliničtí lékaři a vědci se zaměřují jen na krátkodobé výsledky na individuální úrovni a ne na důsledky na úrovni světové populace, které, podle jeho přesvědčení, začnou být brzy evidentní.
Evidentní ve formě změny „vcelku neškodného viru na nezvládnutelné monstrum“.
Jeho obavy se týkají takzvaného ‚imunitního úniku‘. Těm, kteří se potřebují s tématem rychle seznámit, doporučuji přečíst si článek Jemmy Moranové Mutující variace a nebezpečí lockdownů :
Brighteon.com/e86a8f0d-bae2-4c3c-926d-61c60c6e4c38
Pokračujme ale ve čtení stránky.
Bossche uvádí, že několik v současnosti se objevující „mnohem infekčnější“ varianty viru SARS-CoV-2 jsou již příklady „imunitního úniku“ od naší ´vrozené imunity´.
Ty byly nejpravděpodobněji vytvořeny samotnými zásahy vlády; takzvanými nefarmacutickými intervencemi (NPI) – tj. lockdowny a látkovými rouškami. Neoficiálně, ale výstižněji také známými jako nevědecké intervence.
Je přesvědčen, že:
- Pokračující rozjíždění hromadných očkování „s velkou pravděpodobností ještě více zvyšuje ´adaptivní´ imunitní útěk, jelikož žádná ze současných vakcín nezabrání replikaci / přenosu virových variant“.
- A tak, „čím více tyto vakcíny používáme k imunizaci lidí uprostřed pandemie, tím infekčnější se virus stane“.
- A „s rostoucí infekčností přichází zvýšená pravděpodobnost rezistence viru vůči vakcínám“.
Tvrdí, že jeho přesvědčení jsou základní principy, které se studenti učí v prvním semestru vakcinologie – „Profylaktická vakcína by se neměla používat u populací vystavených vysoko infekčnímu tlaku (což je určitě tento případ, kdy v současnosti koluje několik vysoko infekčních variant)“.
Uvádí, že k „úplnému úniku“ tento vysoce mutovatelný virus „potřebuje jen přidat do své oblasti vážící receptor několik dalších mutací“.
Podívejte se jak v tomto aktuálním videu Del Bigtree z Highwire vysvětluje katastrofické důsledky celosvětového prosazování vakcíny:
Brighteon.com/257797f0-06fa-4596-be69-af71bb3adc21
Bossche vysvětluje, že vakcíny na individuální úrovni fungují, ale celkově kvůli „imunitnímu úniku“ vytvářejí nesmírná rizika
Jako zastánce vakcíny je Bossche přesvědčen, že vakcíny na individuální úrovni opravdu fungují a mohou vytvářet imunitu proti zamýšlenému patogenu.
Celkově však masová očkování u velkých populací během pandemie vedou k jevu známému jako „imunitní únik“, což znamená, že virus vytváří varianty, které jsou imunní vůči dostupným vakcínám.
Toto uplatnění přírodního výběru virem vede k ještě nebezpečnějšímu spektru virových kmenů, unikajících z těl hostitelů a vracejících se do „divočiny“, nyní však již nebezpečnější než patogen, který měly vakcíny zastavit.
Ve své prezentaci Bossche varuje, že vakcíny proti covidu „nedokáží zvládat replikaci infekčnějších variant Covidu a mohou dokonce pohánět imunitní únik“.
Vysvětluje, že pro zastavení cyklu mutací a infekcí, který pohánějí „ tři rozličné vlny “ pandemického onemocnění, je rozhodující „vrozená imunita“.
Jenže vrozené imunitě – přirozené imunitě projevované lidmi bez vakcín – nevěnují vakcínami poblázněný zdravotnický establišment a globální politické instituce, které nyní prosazují globální hromadná očkování, ani sebemenší pozornost.
Na následujícím grafu je ve zmíněných 3 vlnách zobrazen vývoj počtu úmrtí na 1000 obyvatel během španělské chřipky v letech 1918-1919:
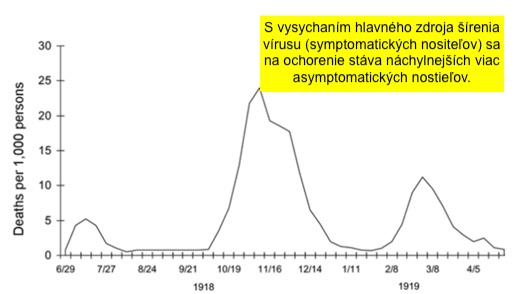
Analýza Dr. Bosscheho se točí kolem role NAC (asymptomatických nositelů) ve zmírňování šíření infekčních virových kmenů.
Jak varuje, když se očkují asymptomatičtí nositelé, vede to k nárůstu infekčnosti viru u symptomatických nositelů.
Řečeno jinak, když hromadně očkujete lidi, kteří nejeví symptomy, vytváříte celo populační tlaky virové adaptace, vedoucí ke zvýšené virální patogenicitě u nositelů, kteří symptomy mají.
Toto pandemii velmi zrychluje a vznikají nebezpečnější kmeny, které jsou stále infekčnější. Jak dále uvádí: „Z toho plynoucí varianty imunitního úniku jsou nyní vůči vakcíně rezistentní.“
To činí vakcínu horší než zbytečnou… ve skutečnosti to urychlilo vývoj superkmenů a zároveň oslabilo následnou imunitní reakci, když se s novými kmeny setkají lidé, kteří byli předtím očkováni.
Naočkování totiž získají silnější specifickou imunitu proti původnímu očkovanému kmeni, ale zároveň jim očkování sníží všeobecnou imunitu proti novým kmenům.
Konkrétně, jak uvádí:
Zvýšené míry infekčnosti vedou u asymptomatických lidí ke zvýšeným mírám přechodné séropozitivity; séropozitivita potlačuje vrozenou imunitu, protože Ag-specifické Ab vytlačují ve vázání ke CoV NAB a brání tréninku vrozeného imunitního systému.
Jinými slovy, vakcína potlačuje imunitní systém a zároveň vytváří smrtící superkmeny .
To vás nutí položit si otázku: Je to všechno záměrné?
„Plně očkováni“ lidé jsou chovateli „superkmenů“ ještě smrtících covidových patogenů
Dr. Bossche dále dokumentuje, že se již objevují důležitá znamení, poukazující na katastrofický výsledek, pokud budou masová očkování pokračovat.
Na dvanácté straně své PDF prezentace uvádí tato „zvláštní pozorování“ ohledně současné pandemie onemocnění Covid-19:
- netypický průběh, resp. vlny pandemie
- objevení se několika mnohem infekčnějších kmenů
- vylučování virů (infekčnějších variant) u plně očkovaných subjektů
To znamená, jak vysvětluje, že vakcína zrychluje adaptivní reakci viru, který vytváří nové kmeny, které jsou mnohem infekčnější a potenicálně smrtící … a že tyto „superkmeny“ se objevují u „plně očkovaných subjektů“.
Tento bod shrnuje prohlášením:
Opatření hromadné kontroly a hromadné očkování u asymptomatických nosičů viru zrychlují VROZENÝ imunitní únik, zatímco hromadné očkování symptomatických nosičů viru zrychluje VROZENÝ a ADAPTIVNÍ imunitní únik.
Jev „adaptivního imunitního úniku“ znamená, že vakcíny vytvářejí na virus přirozené adaptační tlaky, které vedou k tvorbě superkmenů a poté k šíření těmi, kteří již byli očkováni.
Ti lidé však proti těm novým kmenům imunní nejsou , proto se novým supekmenem nakazí i očkováni.
A protože jejich imunitní systémy nikdy nedostaly příležitost aktivně porazit první kmen, mají velmi malou naději na úspěšný boj proti novému superkmeni a mnozí tito lidé zemřou. (To není závěr Dr. Bosscheho, ale Mika Adamse, ale jeho práce tento závěr naznačuje.)
Bosscheho varování:
„Nejnaléhavější nouzovou zdravotní situací mezinárodního rozsahu by se nyní mělo stát okamžité zrušení všech hromadných vakcinačních kampaní proti onemocnění Covid-19“.
Bossche adresoval WHO otevřený dopis, ve kterém varuje, že je třeba okamžitě zastavit kampaň hromadného očkování, jinak lidstvo doplatí na vypuštění „nezvládnutelného monstra“.
V následující části uvádíme překlad tohoto otevřeného dopisu.
Otevřený dopis adresovaný WHO: Okamžitě zastavte všechna hromadná očkování proti Covid-19
Geert Vanden Bossche, DMV, PhD., nezávislý virolog a odborník na vakcíny, bývalý zaměstnanec GAVI a nadace Billa a Melindy Gatesových.
Všem úřadům, vědcům a expertům celého světa, těm, kterých se to týká: všem obyvatelům světa.
Jsem všechno, jen ne antivaxer. Jako vědec obvykle neoslovuji žádnou podobnou platformu o zaujetí postoje v tématech souvisejících s vakcínami.
Jako zanícený virolog a odborník na vakcíny činím výjimku pouze tehdy, když zdravotnické úřady umožňují podávání vakcín způsoby ohrožujícími veřejné zdraví; obvykle tehdy, když se ignorují vědecké důkazy.
K šíření tohoto naléhavého volání mě nutí současná extrémně kritická situace.
Jelikož bezprecedentní rozsah lidského zasahování do pandemie Covid-19 přináší riziko, že povede ke s ničím nesrovnatelné globální katastrofě, tak to hlasitěji a důrazněji už ani vyslovit nelze.
Jak jsem uvedl, nejsem proti vakcinaci. Naopak, mohu vás ujistit, že každá ze současných vakcín byla navržena, vyvinuta a vyrobena vynikajícími a kompetentními vědci. Avšak tento druh profylaktických vakcín je zcela nevhodný, a dokonce vysoce nebezpečný, když se používá v hromadných očkovacích kampaních během virové pandemie.
Vakcinologové, vědci a kliničtí lékaři jsou zaslepeni krátkodobými pozitivními účinky u jednotlivých pacientů, zřejmě je však netrápí katastrofální důsledky pro globální zdraví.
Dokud mi někdo nedokáže, že se po vědecké stránce mýlím, je těžké pochopit, jak současné lidské intervence zabrání kolujícím variantám v tom, aby se změnily na divoké monstrum.
Spěšně dopisuji svůj vědecký rukopis, jehož vydání však bohužel přijde příliš pozdě, vzhledem ke stále narůstající hrozbě rychlého šíření vysokoinfekčních variant. Proto jsem se rozhodl zveřejnit už nyní na LinkedIn souhrn svých zjištění a také své prohlášení na nedávném Vakcínovém summitu v Ohiu.
Minulé pondělí jsem poskytl mezinárodním zdravotnickým organizacím, včetně WHO, svou analýzu současné pandemie na základě vědecky podložených postřehů o imunitní biologii onemocnění Covid-19.
Vzhledem k míře naléhavosti jsem je nabádal, aby zvážily mé obavy a iniciovaly diskusi o škodlivých důsledcích dalšího ‚virového imunitního úniku‘. Pro ty, kteří v tomto oboru nejsou odborníky, připojuji níže přístupnější a pochopitelnější verzi vědeckého zdůvodnění toho záludného jevu.
Přestože není času nazbyt, nedostal jsem zatím žádnou zpětnou vazbu. Experti a politici mlčí, ačkoli očividně stále dychtí mluvit o uvolnění pravidel k zabránění šíření infekce a o „jarní svobodě“.
Moje prohlášení vycházejí výhradně z vědeckých poznatků. Popřít by je měla jen věda.
Přestože člověk může jen sotva vyslovit nějaká nesprávná vědecká tvrzení bez kritiky svých kolegů, zdá se, že vědecká elita, která mometálně řadí našim světovým lídrům, se rozhodla raději mlčet.
Na stůl bylo položeno dostatek vědeckých důkazů. Ty však zůstávají těmi, kdo mají moc jednat, nepovšimnuty.
Jak dlouho může člověk ignorovat problém, když je v současnosti množství důkazů o tom, že nyní ohrožuje lidstvo virový imunitní únik? Sotva můžeme říci, že jsme o tom nevěděli – nebo že jsme nebyli varováni.
Tímto srdceryvným dopisem dávám do sázky celou svou reputaci a důvěryhodnost. Od vás, strážců lidstva, očekávám, že uděláte přinejmenším totéž. Je to nanejvýš naléhavé. Otevřete diskusi. Za každou cenu svraťte tento vývoj!
K vydání globálního varování se připojuje Dr. Vernon Coleman: Vakcíny proti covidu mohou „vyhladit lidskou rasu“
Ke globálnímu varování lidstva se připojuje i známý britský lékař, novinář a publicista Dr. Vernon Coleman.
Ve svém kritickém videu prozrazuje, jak by vakcína proti onemocnění Covid-19 mohla „vyhladit lidskou rasu“:
Autor článku: Mike Adams, Zdroj: naturalnews.com , Zpracoval: Badatel.net
vyšlo 21.3.2021
Související články